
 首页
首页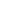 产业动态
产业动态 政策法规
政策法规 企业产品
企业产品 机构信息
机构信息 专题活动
专题活动 报告数据
报告数据 当前位置:
当前位置:
近日,美国洛斯阿拉莫斯国家实验室Jacob S. Spendelow团队和橡树岭国家实验室David A. Cullen团队在阴极催化剂上取得最新进展,作者设计并制备了一种高性能、耐用的聚合物电解质膜燃料电池阴极催化剂,该催化剂由有序的L10-CoPt核和薄Pt壳组成(记为L10-CoPt@Pt-shell)。2022年12月15日,该工作以“Ordered CoPt oxygen reduction catalyst with high performance and durability”为题发表在Cell Press细胞出版社期刊Chem Catalysis上。
中国科技大学谢毅院士、孙永福教授团队针对这项研究成果撰写了Preview文章,以“Higher degree of order enables a more stable fuel cell”为题发表在同日的Chem Catalysis上。
氢燃料电池具有众多优点,比如清洁可持续、能量密度高、能源效率高等,但是其最终应用还面临着重重困难。一个重要挑战就是对高效耐用的阴极氧还原催化剂的需求,这也是学术界和产业界共同努力的目标。
在评估催化剂活性的过程中,使用受控良好的旋转圆盘电极(RDE),新开发的铂基催化剂可以不断地打破电化学氧还原反应(ORR)活性记录,但在更贴近实用的膜电极组件(MEA)测试中,这些高活性却难以实现。RDE和MEA条件之间的巨大性能/认知差距可以归因于明显不同的环境,包括电解质、双电层结构、特定离子吸附、反应物浓度、湿度、温度、离子输送以及气体和水运输的差异。
随着人们对重型车辆应用燃料电池的日益重视,并在其生命周期实现燃料经济性,实现高MEA性能和耐用性比以往任何时候都更加重要。因此迫切需要一种耐用的阴极催化剂,以实现燃料电池和燃料电池车辆的商业化。轻型和重型车辆的耐用性要求分别为5000-8000小时和25000-30000小时,这些耐用性要求对基于无序固溶体Pt-Ni或Pt-Co合金的催化剂构成了巨大挑战,因为从这些合金催化剂中浸出的碱金属可以扩展到整个催化剂颗粒,迅速降低ORR性能。
近日,美国洛斯阿拉莫斯国家实验室Jacob S. Spendelow团队和橡树岭国家实验室David A. Cullen团队在阴极催化剂上取得最新进展,作者设计并制备了一种高性能、耐用的聚合物电解质膜燃料电池阴极催化剂,该催化剂由有序的L10-CoPt核和薄Pt壳组成(记为L10-CoPt@Pt-shell)。在实际的燃料电池膜电极总成测试条件下,阴极催化剂表现出了0.6 A/mgPt的初始质量活性,满足美国能源部的性能目标,同时也满足了经过30000个加速应力测试(AST)电压循环后,质量活性损失小于40%的耐久性目标。L10-CoPt@Pt-shell催化剂具有很高的结构稳定性,作者通过密度泛函理论计算揭示了这种鲁棒性的来源,在有序的CoPt核中观察到Co原子的扩散势垒显著增加。
要点一:通过可规模化的浸渍法制备了金属间ORR催化剂
本研究比较了三种类型的催化剂,包括Pt、合金CoPt和L10-CoPt@Pt-shell颗粒,它们都以碳(Vulcan XC-72)为载体。L10-CoPt@Pt-shell纳米颗粒催化剂是使用Co湿浸渍方法制备的,以商用Pt/C作为起始材料,然后进行高温退火形成有序的L10晶格,再通过酸浸出形成Pt外壳。图1给出了合成的L10-CoPt@Pt-shell纳米催化剂表征结果,经过对晶格条纹和XRD峰的分析,验证了合成的催化剂是CoPt为核,而薄层Pt为壳的结构。
此外,作者还测试了不同样品的Pt L3扩展X射线吸收精细结构(EXAFS)光谱,分析获得的配位数和键长的拟合性清楚地表明,L10–CoPt与PtCo有序分布的颗粒具有相同的Pt-Pt和Pt-Co散射路径,L10-CoPt@Pt-shell表现出更多的Pt-Pt配位,但是减少了Pt-Co配位,与预先浸出Co的表面增加了Pt-Pt键一致。与L10–CoPt相比,L10-CoPt@Pt-shell中的Pt-Pt键长增加也验证了催化剂颗粒近表面Co的缺失。
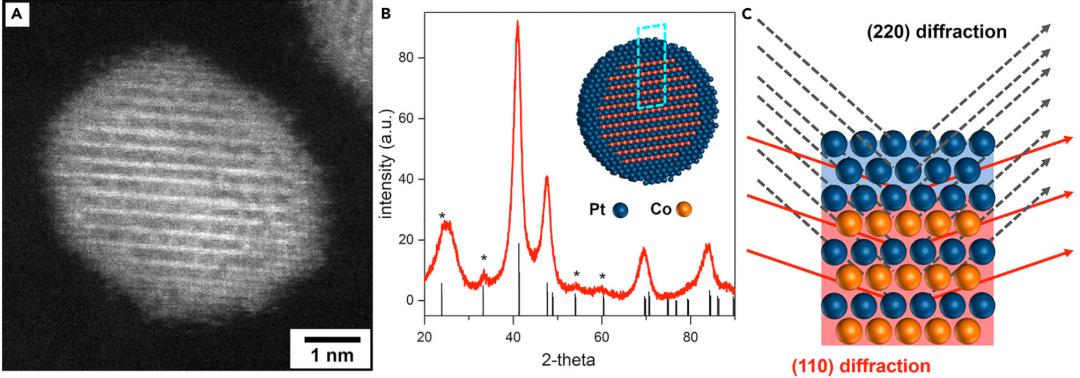 图1 L10-CoPt@Pt-shell纳米催化剂的表征和分析
图1 L10-CoPt@Pt-shell纳米催化剂的表征和分析
要点二:在燃料电池测试中实现了0.6 A/mgPt的质量活性和高耐用性
接下来,根据能源部测试协议,作者在150 kPaabs H2/O2和0.9 V下测试三种催化剂的质量活性。合金CoPt和L10-CoPt@Pt-shell在寿命初期(BOL)的质量活性为0.60 A/mgPt,远高于能源部0.44 A/mgPt的目标,相似的性能可归因于表面Pt原子有相似的粒度和相似的压缩应变。相比之下,Pt催化剂的BOL质量活性为0.29 A/mgPt。在30000个AST周期之后,L10-CoPt@Pt-shell催化剂的质量活性为0.36 A/mgPt,比合金催化剂(0.27 A/mgPt)要高得多。30000个AST周期后的质量活性损失达到了能源部要求的耐用性目标(40%或更少)。相比之下,合金催化剂遭受了56%的质量活性损失。
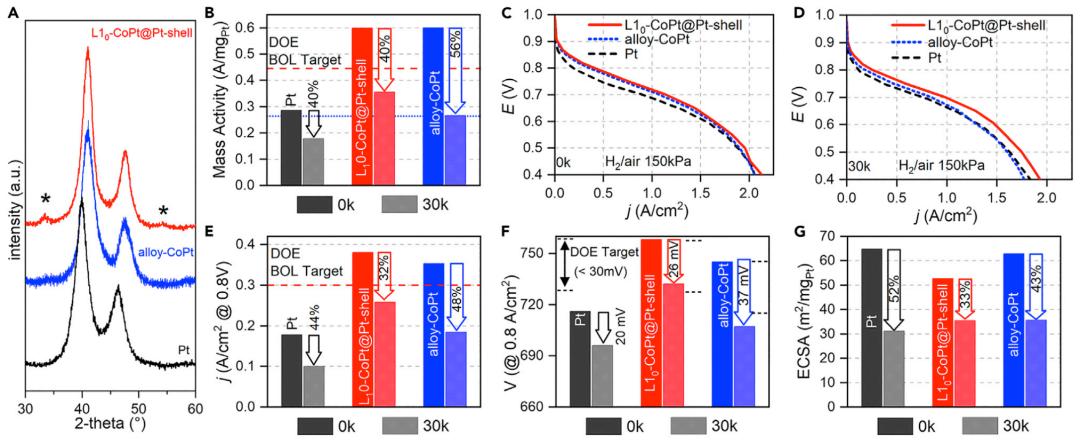 图2 碳负载Pt、CoPt合金和L10-CoPt@Pt-shell催化剂的晶体结构信息和燃料电池测试结果
图2 碳负载Pt、CoPt合金和L10-CoPt@Pt-shell催化剂的晶体结构信息和燃料电池测试结果
要点三:在0.67 V时,0.1 mgPt/cm2催化剂的H2/空气功率密度为0.87 W/cm2
图2C和2D显示了在BOL和30000个AST周期后(即寿命终止,EOL)的H2/空气下测试的三种催化剂的极化曲线。图2C中,L10-CoPt@Pt-shell催化剂在动力学和欧姆区域都表现出最高的性能,在0.6 V时电流密度为1.63 A/cm2,在0.67 V的热抑制极限下为1.30 A/cm2,功率密度为0.87 W/cm2。当在250 kPaabs测试时,L10-CoPt@Pt-shell的性能为1.1 W/cm2,远远超过了能源部1 W/cm2的目标。
在BOL上,两种合金催化剂表现出类似的性能,与BOL质量活性测试结果非常吻合。30000个AST周期后,L10-CoPt@Pt壳和CoPt合金催化剂的极化曲线之间的差异变得更加显著,与质量活性测试中观察到的趋势相匹配。与合金催化剂相比,L10-CoPt@Pt-shell催化剂具有更高的耐用性,在30000个AST周期后,在0.6V的热抑制极限下显示1.49 A/cm2的电流密度。除了H2/O2测量的质量活性和耐用性目标外,能源部还设定了在0.8 V和0.8 A/cm2处H2/空气的性能和耐久性目标。在BOL,L10-CoPt@Pt-shell催化剂的电流密度为0.38 A/cm2,远远超过能源部0.3 A/cm2的目标,催化剂AST后仅损失32%,而合金CoPt的损失为48%。L10-CoPt@Pt-shell催化剂在0.8 A/cm2的电压损失仅为26 mV(758-732 mV),满足了DOE 30 mV的目标,而合金CoPt电压损失为37 mV。L10-CoPt@Pt-shell的33%的ECSA损失也是所有测试催化剂中最轻微的。
 图3 L10-CoPt@Pt-shell催化剂在BOL和EOL中的表征
图3 L10-CoPt@Pt-shell催化剂在BOL和EOL中的表征
之后,作者试图从材料角度破译L10-CoPt@Pt-shell的高性能和耐用性的起源。HAADF-STEM显微照片中观察到,催化剂在30000个催化剂AST周期后保持了清晰的L10-CoPt@Pt-shell结构,Co损失较少,这说明催化剂的结构得到了很好的保持,这也得到了XRD和X射线荧光光谱的验证。
要点四:利用DFT评估了依赖有序性的空位介导扩散动力学
为了进一步解释实验观察到的L10-CoPt@Pt-shell催化剂的高耐用性,使用DFT原子模型系统地研究了杂乱程度不断增加的CoPt体相内的Co稳定性。在催化表面,像Co这样的非贵金属元素的损失主要归因于在燃料电池中的溶解。对于含有Pt壳的核壳结构,假设表面只有Co原子能溶解到溶液中,溶解必须伴随着内部扩散,以便随着时间的推移进一步降解。虽然固体中的扩散可以通过间隙、集体或空位介导机制进行,但空位介导机制被认为是类似尺寸原子在金属和合金中扩散的主导机制。
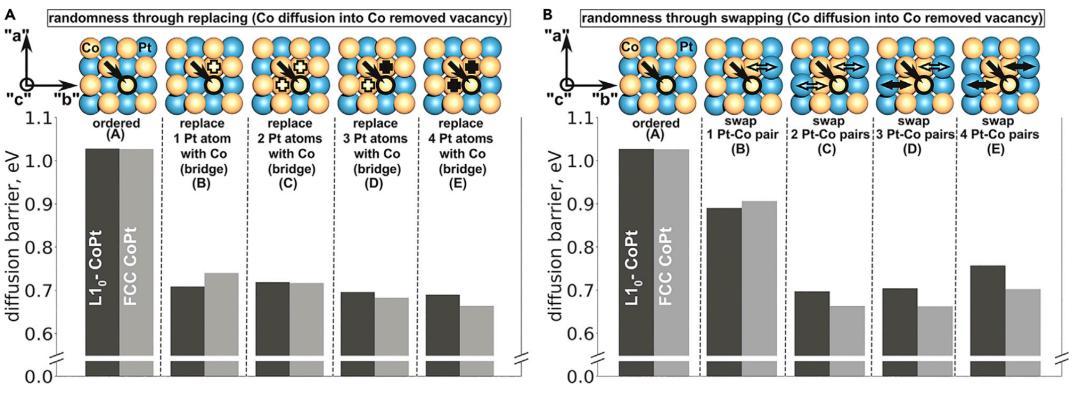 图4. DFT计算当原子排列变得无序时,Co原子的扩散势垒
图4. DFT计算当原子排列变得无序时,Co原子的扩散势垒
为了研究随机性增加对核内Co稳定性的影响,因此考虑了一个空位介导的扩散模型来计算Co扩散到空位的扩散能垒,如图4所示。作者假设,局部环境中的无序程度或浓度会影响扩散能量,从而随着时间的推移导致核退化。通过两种不同的方法,即替换和交换,在局部环境中引入系统随机性,研究了扩散能垒的变化。
结果表明,高有序性对于确保Co更高的动力学稳定性至关重要。同样,对于通过Pt空位的Co扩散,作者观察到,与有序结构相比,无序结构可能导致较低的Co扩散能垒。这些计算结果与实验观察结果非常一致,即L10-CoPt比fcc-CoPt更有效地稳定Co。
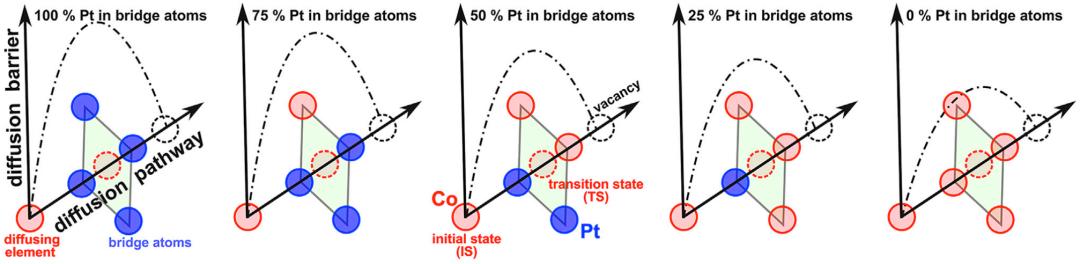 图5. 假设的扩散能垒随着局部环境浓度的变化或无序程度的增加而变化
图5. 假设的扩散能垒随着局部环境浓度的变化或无序程度的增加而变化
总结
通过优化核壳结构的CoPt催化剂的晶体有序性和尺寸分布,作者设计并制备了符合能源部燃料电池催化剂和MEA目标的ORR催化剂。制备的亚-4 nm催化剂由一个有序的L10核和Pt壳组成。该催化剂在MEA测试条件下表现出出色的性能和耐用性。高性能归功于ORR动力学得到增强,因为表面Pt壳施加了压缩应变,以及亚-4 nm催化剂颗粒暴露的相对较大的ECSA。高耐用性源于有序金属间结构提供的出色的Co稳定,它能够实现稳定的核壳结构,同时保留晶格收缩和颗粒尺寸。
专家述评
中国科技大学谢毅院士、孙永福教授团队对这篇研究成果进行了专业点评,详情如下。
Pt基电化学氧还原反应催化剂往往展现出优异的活性,但稳定性不足。在本期Chem Catalysis中,来自美国橡树岭国家实验室的Cullen以及洛斯阿拉莫斯国家实验室的Spendelow等人提出了一种具有高有序度的PtCo@Pt核壳结构催化剂,在实际的聚合物电解质膜燃料电池(PEMFC)中实现了更优的性能,并拥有长效的耐久性。
“绿氢”的有效利用对可持续发展的未来至关重要。将氢的电化学氧化(HOR)和氧气的电化学还原(ORR)相结合的燃料电池技术,是一项有望实现这一目标的技术。燃料电池装置的商业可行性主要受制于具有较高热力学能垒的阴极ORR。在这个过程中,分子氧(O2)将被催化剂上的四个电子和质子选择性地激活并还原,生成水。由此考虑,开发具有优异活性和长期耐久性的新型ORR催化剂通常有助于提高整个装置的效率。
在过去数十年中,Pt基纳米材料是所有催化材料中作为ORR电催化剂性能最优越、最有希望的。然而,纯Pt催化剂的活性和耐久性依旧欠缺,其成本和商业可行性仍阻碍了实际运用。针对这一问题,DFT计算得到了催化活性和关键反应中间体的吸附能量之间存在“火山型”关系,这表明通过改变Pt以及其他贱金属的电子结构可实现出色的性能。在这方面,通过形成Pt-M(M为过渡金属)合金以调控Pt的结构,有望优化其电催化性能同时大大减少阴极中昂贵的Pt的使用量。对于常见的无序的或固溶体PtM合金,贱金属可能从这些催化剂中不断脱落,导致其ORR性能的迅速下降(图1A)。要使催化剂能够在更具腐蚀性的MEA操作条件下工作更长时间,Pt和贱金属之间更强的键合作用,以及可增加贱金属扩散障碍的原子尺度的有序性十分关键。
基于此,来自美国橡树岭国家实验室的Cullen以及洛斯阿拉莫斯国家实验室的Spendelow等人利用PtCo@Pt核壳结构金属间化合物催化剂中的高度有序性,使得金属键具有类似共价键的特征。
研究人员使用商业Pt/C催化剂为原材料,先进行Co湿法浸渍,随后高温退火得到金属间有序的PtCo@Pt核壳催化剂。高角环形暗场-扫描透射电镜表征显示了催化剂的核壳结构(图1B和1C),其中有序PtCo金属间化合物核的直径约为3.5纳米,Pt薄壳的厚度约为0.4纳米。由无序至有序的结构变化被认为有望提升燃料电池中电催化剂的活性和耐久性。将合成所得的PtCo@Pt催化剂装载于MEA配置的气体扩散电极上以研究组成效应对其性能和耐久度的影响。在0.9V和150 kPaabs氢气/氧气条件下,其初始阶段质量活性可达到0.6 A/mgPt,远高于美国能源部0.44 A/mgPt的目标(图1 D)。在30000次循环的加速应力测试后,PtCo@Pt催化剂能展现出0.36 A/mgPt的质量活性,显著高于合金PtCo(约0.27 A/mgPt)以及商业Pt催化剂(约0.18 A/mgPt)。循环测试后该催化剂约损失40%的质量活性,仍可满足美国能源部耐久性目标。作为比较,无序合金PtCo催化剂损失了56%的催化质量活性。更为重要的是,PtCo@Pt催化剂在动力学以及欧姆区均展现出最高的活性和耐久性,0.6V条件下30000次循环后电流密度为1.49 A/cm2(图1E)。
进一步地,研究人员通过细致的探究揭示了PtCo@Pt催化剂高耐久性的来源。高空间分辨率STEM-EDS元素分析结果显示,反应后材料晶格仅仅发生由于Co析出的轻微扩张,核壳结构整体得以保持(图1F)。X射线荧光谱进一步证明了相比于其他两种合金PtCo催化剂,PtCo@Pt催化剂反应后的Co损失最小,因此具有最高的耐久性(图1G)。
此外,研究人员通过DFT计算系统性探究了PtCo中Co稳定性与无序度之间的关系(图1H)。首先,贱金属Co的损失主要是由于苛刻的燃料电池测试条件导致的溶解。对于PtCo@Pt的核壳结构,假设仅有表面的Co原子能溶出,必然伴随着内部扩散以及随后的内部结构随时间的破坏。进而,研究人员提出了一种空位介导的机制以厘清扩散能量以及无序度存在的关系。结果表明,有序结构中Co的扩散能垒最高,表明高度的有序性对动力学稳定性至关重要。计算结果与实验结果很好地吻合,PtCo@Pt核壳结构相较于fcc-PtCo能更有效地稳定Co原子。
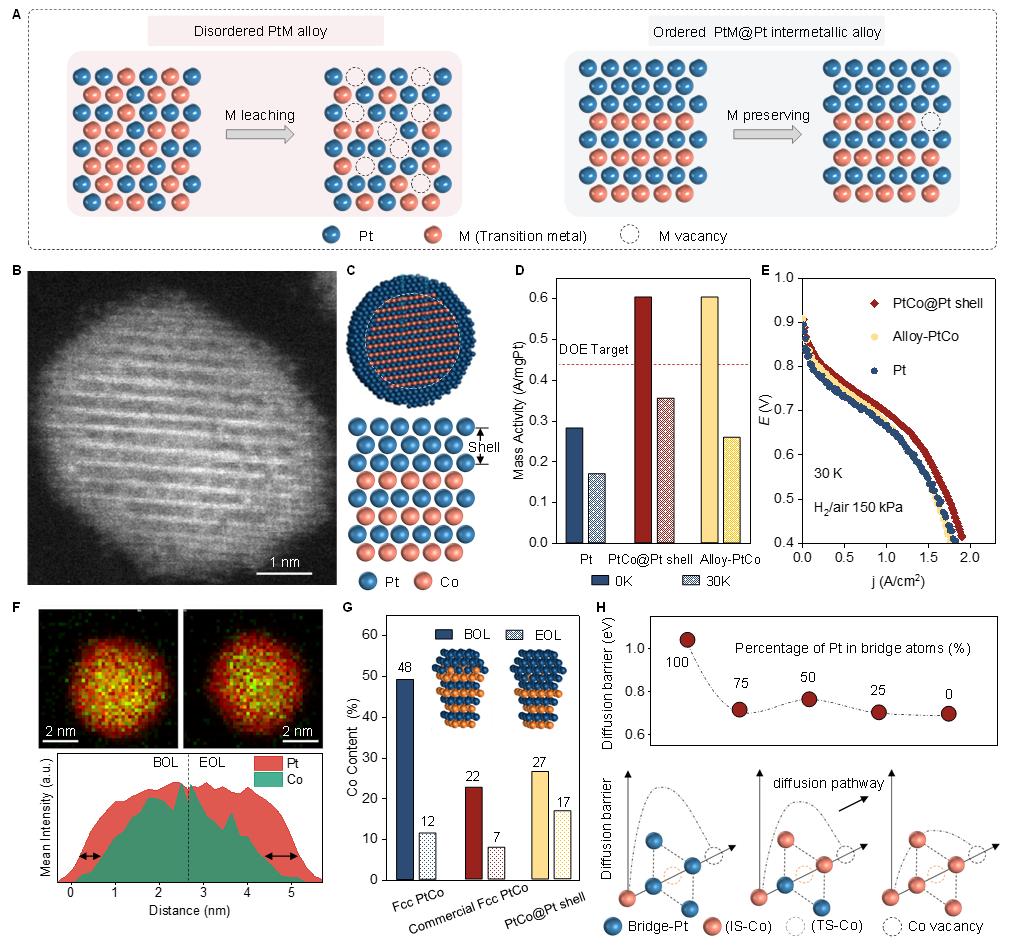 图1 PtCo@Pt核壳金属间化合物催化剂中的高度有序性造就了更稳定的燃料电池
图1 PtCo@Pt核壳金属间化合物催化剂中的高度有序性造就了更稳定的燃料电池
总之,耐用的Pt基催化剂对燃料电池设备至关重要,特别是需要考虑生命周期成本和经济性问题时。核壳结果催化剂已被证明有利于这一目标的实现。该工作不仅在实验上开发了一种性能优异的催化剂,而且在理论上证实了“催化剂中高度有序性”概念可赋予催化剂更好的耐久性。尽管在催化剂研发上取得了令人激动的进展,但包括三相界面、传质以及测试方法在内的问题仍需解决,以实现真正的商业应用。
相关论文信息
论文原文刊载于CellPress细胞出版社旗下期刊Chem Catalysis上,
▌论文标题:
Ordered CoPt oxygen reduction catalyst with high performance and durability
▌论文网址:
https://www.cell.com/chem-catalysis/fulltext/S2667-1093(22)00607-8
▌DOI:
https://doi.org/10.1016/j.checat.2022.10.030
 大连化物所开发煤化工废水制氢联产淡水中试装置开车成功
大连化物所开发煤化工废水制氢联产淡水中试装置开车成功 热门动态
热门动态 推荐活动
推荐活动



 新闻排行榜
新闻排行榜




